What is Fabry's disease?
Fabry disease was first described in 1898 by 2 doctors: Dr. William Anderson in England and Dr. Johannes Fabry in Germany. Hence sometimes the name "Anderson-Fabry disease" is used.
Fabry disease is a hereditary disorder affecting approximately 2,000 to 4,000 people worldwide. It occurs in both men and women and is one of the lysosomal storage diseases. These diseases are characterized by a shortage of enzymes.
Fabry patients lack an important enzyme called α-galactosidase A, or it is only produced in very small quantities. Without this enzyme, certain substances or waste are not removed from the body but accumulate in cells in the main organs of the body, including the kidneys, the heart and the central nervous system. This gradual accumulation of waste products disrupts the normal functioning of the cells and causes Fabry's disease.
About the Fabry enzyme
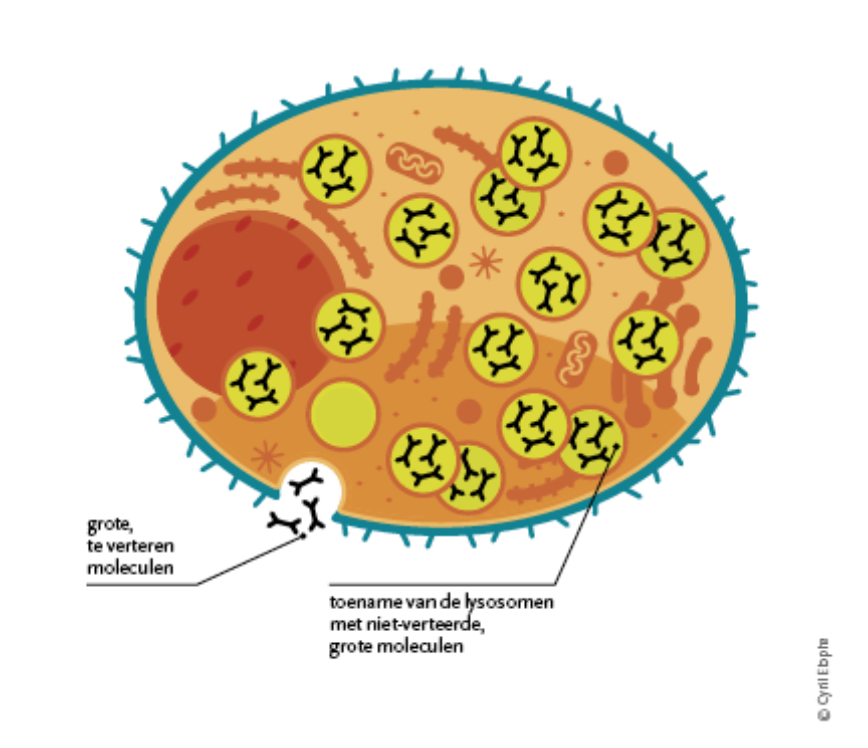
The enzyme α-galactosidase A is only one of a series of enzymes that are normally present in the lysosome. Lysosomes are small "recycling centers" in our cells that play an important role in the degradation of waste to smaller substances that can be reused by the cell or that are excreted from the body.
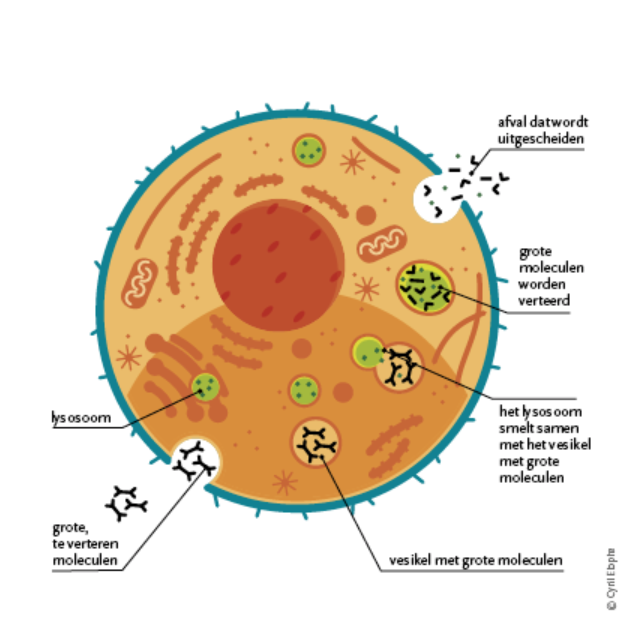
The lack of this enzyme leads to an accumulation of waste substances, lipids (fat structures) in the cells. The main waste product is globotriaosylceramide or GL-3.