

Laboratory tests used during diagnosis
To find out if someone has Fabry disease, tests need to be done. These tests could include:
Enzyme activity tests
One of the tests used to further investigate the suspicion of someone having Fabry disease is to analyse alpha-GAL A enzyme activity from a blood sample. In people with Fabry disease, the activity level of this enzyme is typically (much) lower than people without Fabry disease. Testing enzyme activity can also be done on other sample types such ‘Dried Blood Spots’ (DBS) where small amounts of blood are dried onto filter paper. The levels of enzyme activity can be a good indicator if the disease is present or not. However, because of the nature of the disease and how it is inherited (via the X-chromosome), in some cases this test in not sufficient on its own. Genetic tests are often performed to confirm or exclude the diagnosis of Fabry disease.
Genetic tests
Genetic tests can be performed to confirm or exclude the presence of a Fabry disease causing mutation, as well as to find out what genetic defects are causing the disease. This is called ‘genotyping’ and can give useful genetic information to doctors.
A person with Fabry disease and their family may be offered counselling at this stage so they can learn about how this will impact their lives.
Because Fabry disease can affect different organ systems, doctors may perform other tests and examinations to see how Fabry disease is affecting these different parts of the body.
People with Fabry disease may manifest overt Fabry disease symptoms during the first decades of life but an accurate diagnosis is very important so they can be monitored for symptoms over time. Also a definitive diagnosis can help to find any other family members who may have the disease and need treatment.
Is there a treatment for Fabry disease?
People with lysosomal storage diseases cannot produce enough specific enzymes to make the body work properly. These enzymes, which cause the breakdown of a large number of different types of substances, are not present at all or only in very small quantities.
The diagram below explains the processes behind a lysosomal storage disorder.
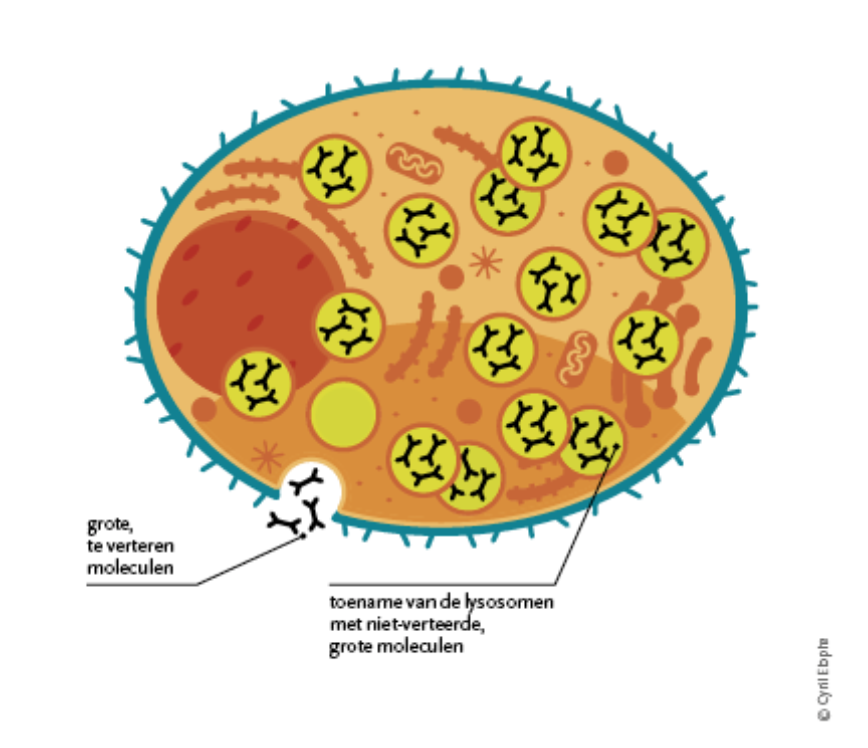
The result is an accumulation of material that disrupts the normal functioning of the cells. As a result, blood vessels are affected, causing damage to important organ systems, which in turn leads to life-threatening problems.
In Fabry disease, the body is unable to produce the enzyme α-galactosidase that is needed to excrete certain waste products. Since 2001, enzyme replacement therapy has been available.
Enzyme replacement therapy (Enzyme Replacement Therapy or ERT)
With this therapy, the missing lysosomal enzyme is administered by means of infusions with an enzyme that is very similar to the missing human enzyme.
This type of treatment has been available for the treatment of Gaucher disease for more than 18 years and has recently been developed for a number of other lysosomal storage diseases (Pompe disease, Hurler / Scheie syndrome).
The treatment consists of regular infusions of the missing enzyme. This makes the metabolism (metabolism) normal again and stops the accumulation of the waste.
The diagram below explains the effect of enzyme replacement therapy
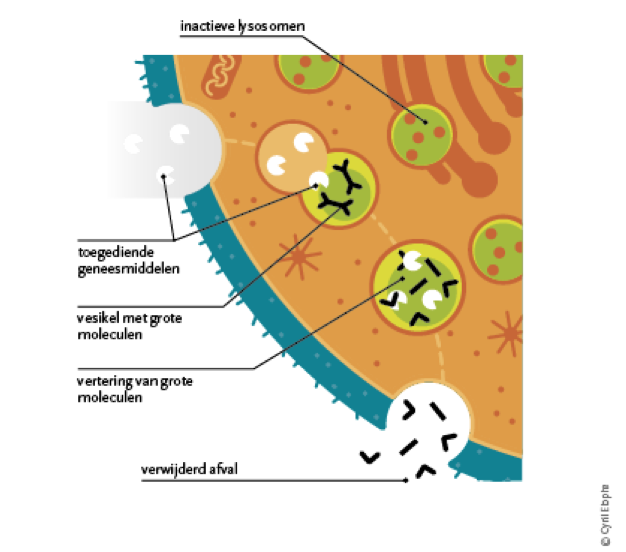
Enzyme replacement therapy for Fabry disease is given every 2 weeks.
Fabry disease is a progressive disease and there is increasing evidence that the sooner after the first symptoms appear, the therapy starts, the better the results will be. The therapy is most likely to succeed if it is started at the moment that there are no late complications and on the condition that the enzyme replacement therapy is administered regularly (every 2 weeks).
Thanks to the therapy, the course of the disease can be delayed or changed and late complications can be avoided, so that many patients can lead a normal life. However, irreversible damage to the organs cannot be remedied with enzyme replacement therapy.
Family testing
Because Fabry disease is an X-linked disease, it is likely that other family members of an affected individual have Fabry disease too. Family screening enables a doctor to diagnose and monitor family members with Fabry disease properly and manage the disease.
‘Family screening’ is a way to find out if other family members are affected by the disease as well. If family members want to participate in family screening, a genetic and/or enzyme activity test should be done from a blood sample. This test will determine whether family members are also affected by Fabry disease. Patients can help their doctor by making a ‘family tree’ with the names of their family members including grandparents, aunts, uncles and cousins.
For those family members who have the faulty gene, but no health problems at the moment, the doctor can regularly monitor them for possible development of signs or symptoms of Fabry disease and give them appropriate care if such signs/symptoms appear. For those family members who do not have the faulty gene, it can bring peace of mind.
The process of family screening can be very emotional for family members and should be managed by a trained heath care professional or genetic counsellor. They can help:
- Research family history
- Provide information and support families to make decisions about testing and treatment
- Listen and advise if there are any worries about the process
- Help to bring in contact with patient associations





